![]()
Overview
Gene prioritization at human GWAS loci is challenging due to linkage-disequilibrium and long-range gene regulatory mechanisms. However, identifying the causal gene is crucial to enable identification of potential drug targets and better understanding of molecular mechanisms. Mapping GWAS traits to known phenotypically relevant Mendelian disease genes near a locus is a promising approach to gene prioritization. MendelVar is a novel web-based platform to support gene prioritization using data from Mendelian disease genes, variants identified in clinical genetics and data from disease ontologies.
The platform
MendelVar, presented in this Bioinformatics paper, provides a quick overview of possible impact of Mendelian disease-related genes on user’s complex phenotype of interest. It returns the details of all known broadly defined Mendelian diseases and their causal genes found in the custom genomic intervals as well as overlapping pathogenic rare mutations responsible for Mendelian disease. Enrichment of Disease Ontology, Human Phenotype Ontology terms among the Mendelian genes gives the researcher an overview of any shared features with their trait of interest, e.g. in terms of anatomy.
Data sources
MendelVar uses all the confirmed gene-disease relationships featured in OMIM and complements it with three more specialist data sources for Mendelian disease: Orphanet (a database centred on rare, typically monogenic disease), expertly curated gene panels used for diagnostics from Genomics England PanelApp and results from the on-going Deciphering Developmental Disorders Study (made available in DECIPHER). MendelVar includes short disease descriptions sourced from OMIM, Orphanet, Uniprot and DO. In addition, MendelVar cross-references input genomic intervals against ClinVar.
Usage
MendelVar accepts a user defined list of genomic intervals or a list of top SNPs. Top SNPs can be used to create genomic intervals in two ways in MendelVar: using pre-set basepair flanks or via generation of individual LD-based boundaries around each top SNP, identified either through dbSNP rsIDs or positional coordinates. The LD-based intervals can be also optionally extended to the nearest recombination hotspot. There is full support for hg19/GRCh37 and hg38/GRCh38 human genome builds for input. Six 1000 Genomes populations - EUR, CEU, AFR, AMR, EAS, SAS supported via LDlink for LD-based genomic interval generation.
The web-browser will return a compressed file containing the results of the enrichment and annotation analyses. An online tutorial provides clear documentation on how to use the platform and how to interpret the results.
See Figure 1 for an illustration of the different user routes through MendelVar.
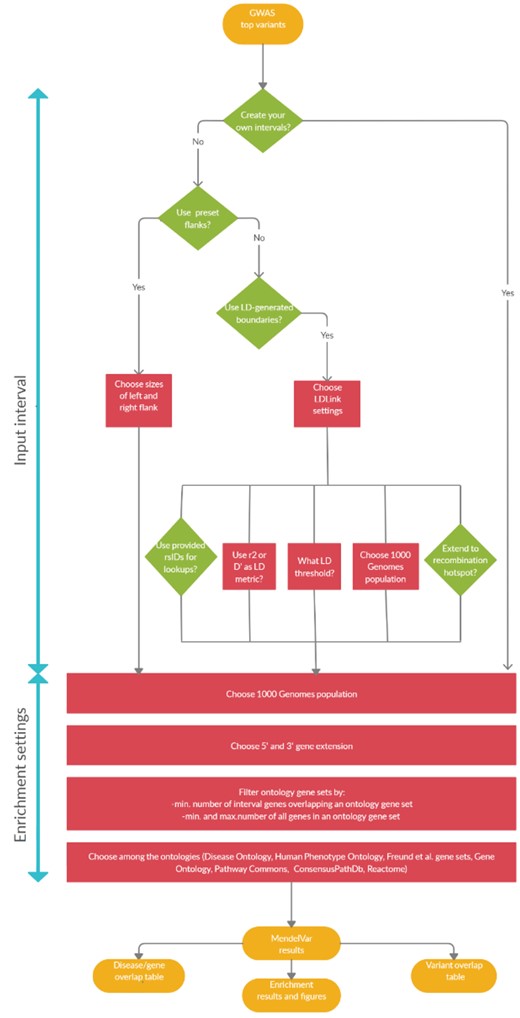 Figure 1: A flowchart demonstrating three possible user routes through MendelVar: (a) left: MendelVar generates fixed genomic intervals using preset left and right flanks against a user-submitted list of genomic positions; (b) centre: MendelVar generates flexible genomic intervals using LD pattern in the region around each user-submitted position/variant rsID; (c) right: MendelVar accepts user-submitted genomic intervals. The genomic intervals generated or obtained from user are subsequently bisected with coordinates for genes and variants known to cause Mendelian disease. Ontology terms associated with Mendelian disease in HPO, DO are propagated to causal genes and are tested for enrichment among target genes in input genomic intervals. MendelVar also provides an option for enrichment testing with Gene Ontology and biological pathway databases.
Figure 1: A flowchart demonstrating three possible user routes through MendelVar: (a) left: MendelVar generates fixed genomic intervals using preset left and right flanks against a user-submitted list of genomic positions; (b) centre: MendelVar generates flexible genomic intervals using LD pattern in the region around each user-submitted position/variant rsID; (c) right: MendelVar accepts user-submitted genomic intervals. The genomic intervals generated or obtained from user are subsequently bisected with coordinates for genes and variants known to cause Mendelian disease. Ontology terms associated with Mendelian disease in HPO, DO are propagated to causal genes and are tested for enrichment among target genes in input genomic intervals. MendelVar also provides an option for enrichment testing with Gene Ontology and biological pathway databases.
Paper
‘MendelVar: gene prioritization at GWAS loci using phenotypic enrichment of Mendelian disease genes’ by M K Sobczyk, T R Gaunt and L Paternoster in Bioinformatics (2021).